Lysosomale Speicherkrankheiten (Lysosomal Storage Disorders, LSD) sind eine Gruppe von mehr als 50 seltenen erblichen Stoffwechselkrankheiten. Die Krankheiten sind gekennzeichnet durch eine abnorme Anhäufung verschiedener toxischer Stoffe in den Körperzellen als Folge von Enzymmängeln.
Lysosomale Speicherkrankheiten betreffen das Lysosom, eine Struktur in den Zellen, die Stoffe wie Proteine, Kohlenhydrate und alte Zellteile abbaut, damit der Körper sie wiederverwerten kann. Infolgedessen können verschiedene Teile des Körpers betroffen sein, darunter das Skelett, das Gehirn, die Haut, das Herz und das zentrale Nervensystem. Neue lysosomale Speicherkrankheiten werden weiterhin identifiziert.
Bei der Diagnose einer Speichererkrankung muss sich mit vier Fragen beschäftigt werden:

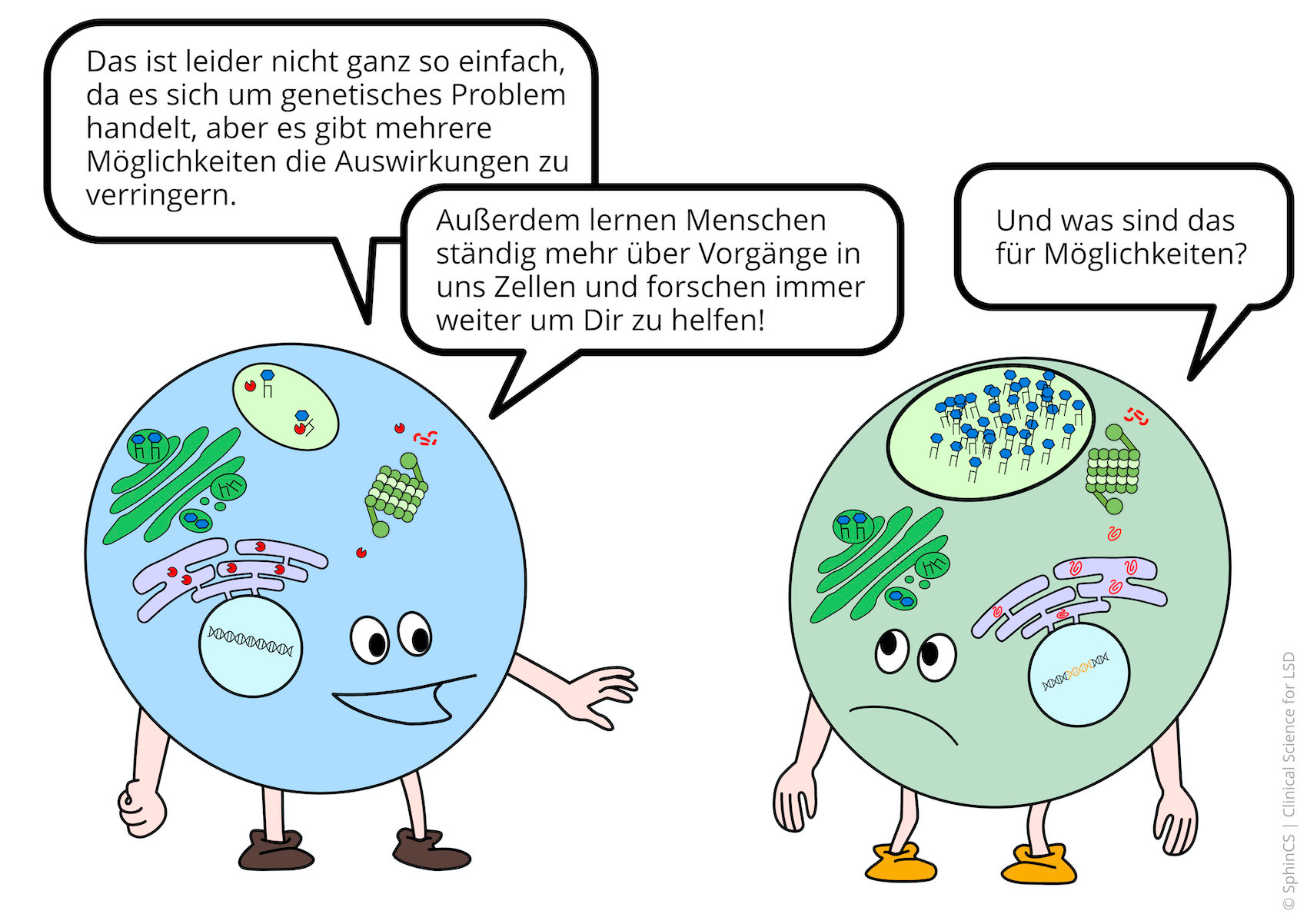
Behandlung nicht möglich, selten, komplex, schwer zu verstehen, schlechte Prognose, … das sind die Stichworte, die aus dem Medizinstudium haften geblieben sind. Doch wenn wir aktuell 2020 die aktuelle Forschung zusammenfassen, dann gibt es bereits wirksame und zugelassene Therapien und weitere Therapieansätze machen Hoffnung.
Die Behandlung des Zentralen Nervensystems ist eine besondere Herausforderung, die durch Substratinhibitation und Gentherapie sich weiter entwickelt
Da immer besser verstanden wird, was in den Zellen und in den Organen bei lysosomalen Erkrankungen passiert, erarbeitet die Grundlagenforschung immer neue Ansatzpunkte für Therapien
1983 wurde erstmals eine Knochenmarkstransplantation bei einer lysosomalen Erkrankungen durchgeführt. Transplantiert wurde in London durch Dr. Hobbs ein Junge mit MPS 1. Es hat sich nicht bewahrheitet, dass alle lysosomalen Erkrankungen so behandelt werden können. Es sind die schweren Verlaufsformen einiger weniger Erkrankungen, wie MPS 1 und M. Farber, bei denen die Knochenmarkstransplantation auch 2020 noch die Therapie der Wahl ist.
1991 ging ein Raunen durch den medizinischen Blätterwald. Roscoe Brady und sein Team vom NIH hatten 12 Gaucher-Patienten in einer Studie genau mit dem Enzym behandelt ( Glucocerebrosidase ), welches bei dieser Erkrankung fehlt. Das Enzym wurde 14tägig über Infusionen gegeben. Die Krankheitssymptome Milzvergrößerungen Mangel an Blutplättchen und Blutungsneigung sprachen hervorragend auf die Therapie an. Damals wurde das Enzym aus Mutterkuchen gewonnen, heutzutage wird es in größeren Mengen und sicherer biotechnologisch hergestellt.
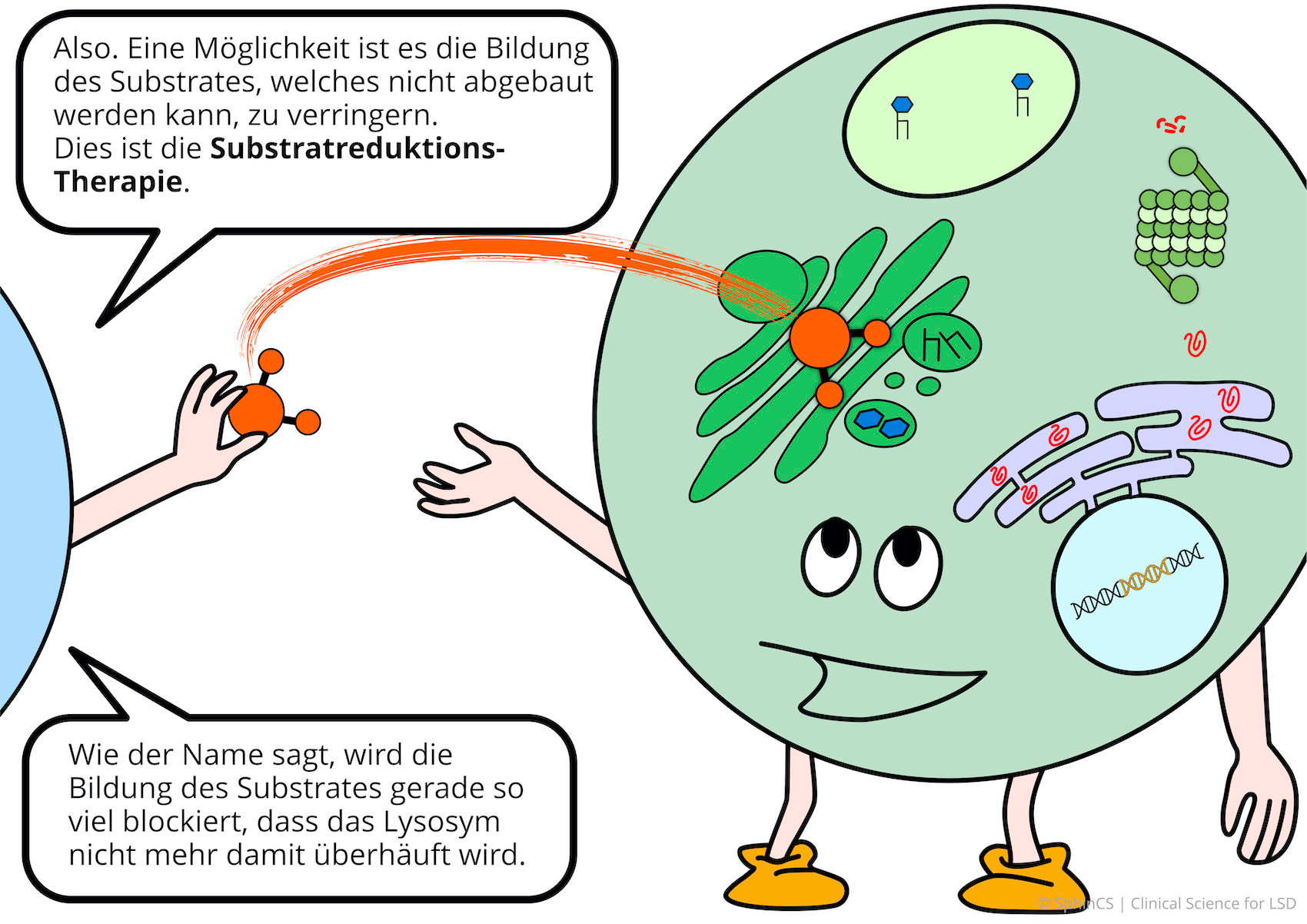
Das Therapieprinzip beruht darin, dass die Substanz die nicht abgebaut werden kann (=Substrat), nur noch sehr begrenzt gebildet wird. Man hemmt das Substrat aufbauende Enzym, dann fällt der Mangel an dem Enzym welches das Substrat abbbaut kaum ins Gewicht.
Chaperone binden spezifisch an fehlerhaft gefaltete Enzyme und stabilisieren die Struktur des Enzyms. An der Art der Mutation kann erkannt werden, ob eine Mutation eine fehlerhafte Fältelung verursacht. Patientienten mit solchen Mutationen kommen für eine Chaperontherapie in Frage.
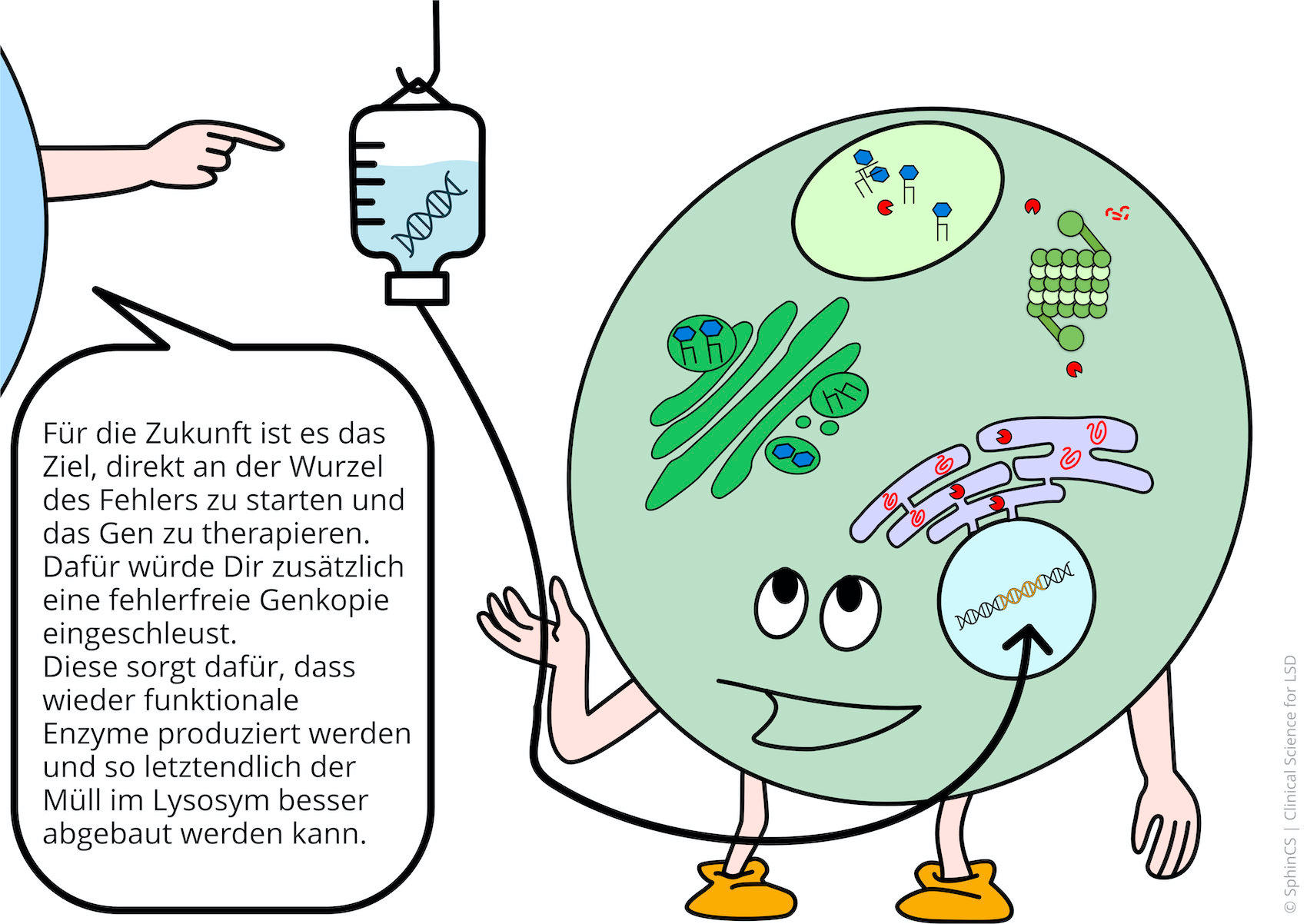
Gentherapien heilen Erbkrankheiten durch das Einschleusen einer fehlerfreien Genkopie. Hört sich einfach an, ist aber höchst kompliziert. Das Einschleusen gelingt durch virale Vektoren. Insbesondere erfolgreiche Grundlagenforschung mit dem AAV9-Vektor, der das gesunde Gen in Nervenzellen einbringt, führte in den letzten beiden Jahren zu der Entwicklung von zahlreiche Therapiestudien für lysosomale Erkrankungen.
Für eine Übersicht der anderen Art, laden wir Sie ein sich den folgenden Comic anzuschauen. Klicken bzw. Tippen Sie einfach auf das erste Bild oder laden Sie sich den Comic als PDF herunter.

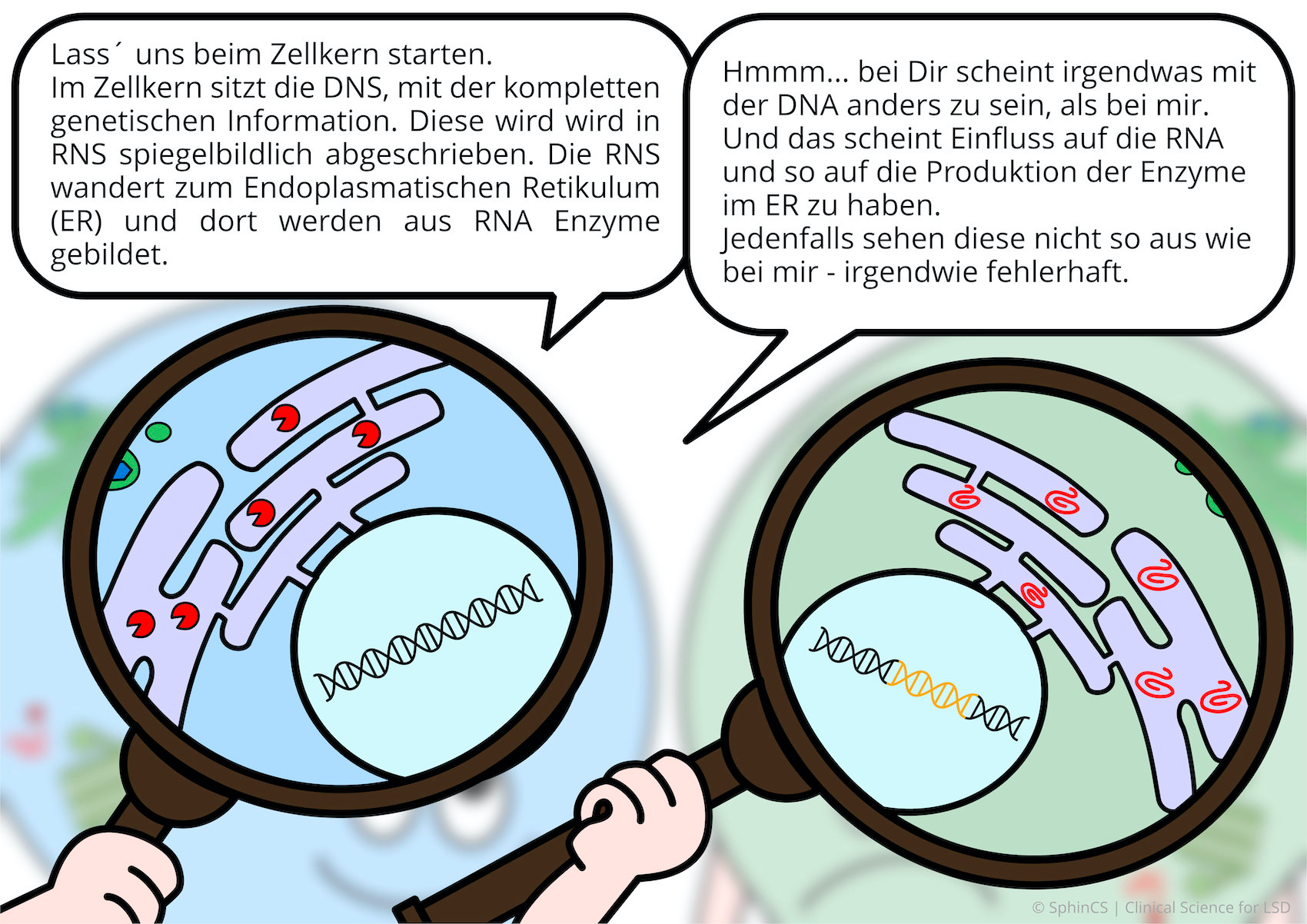
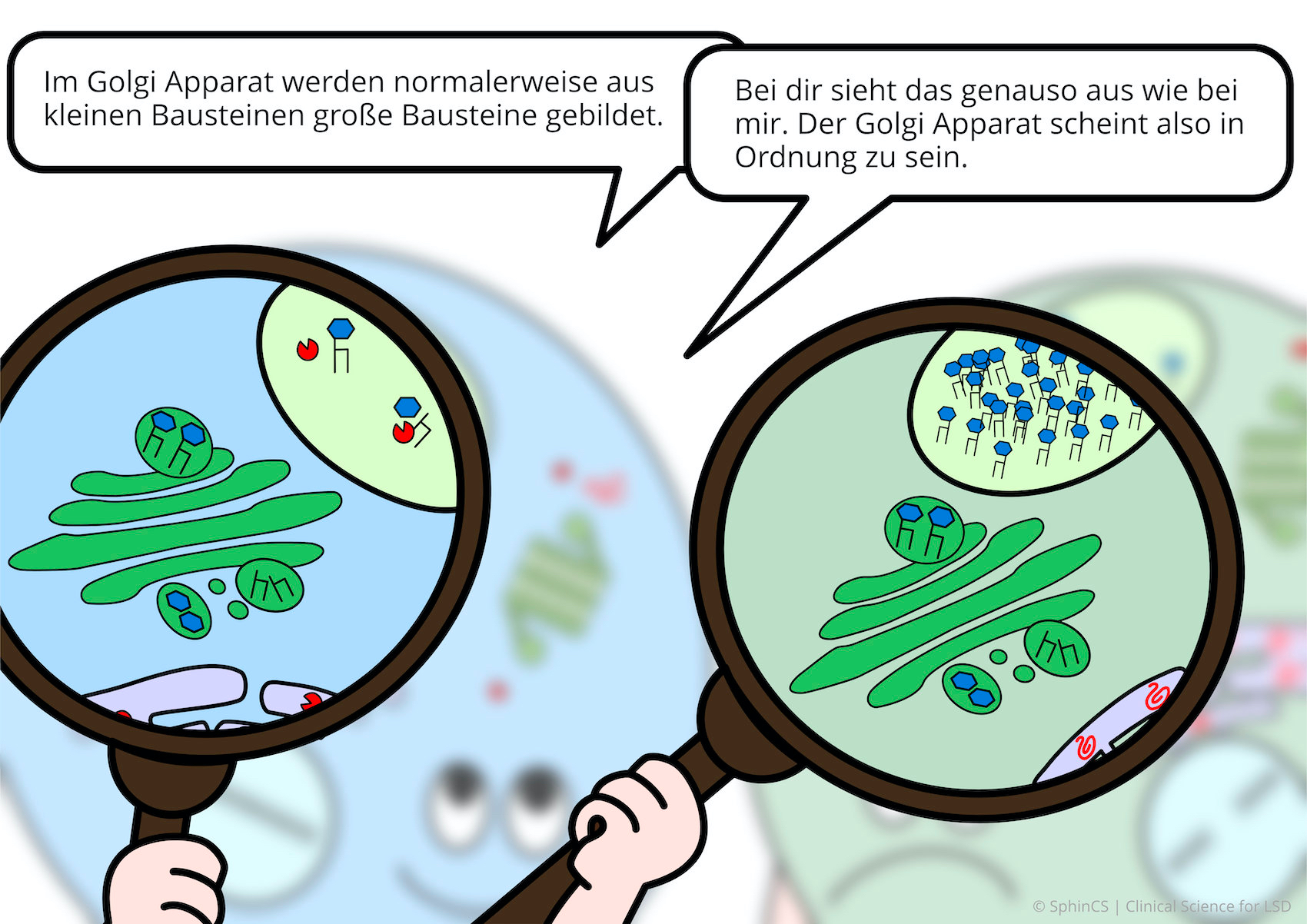
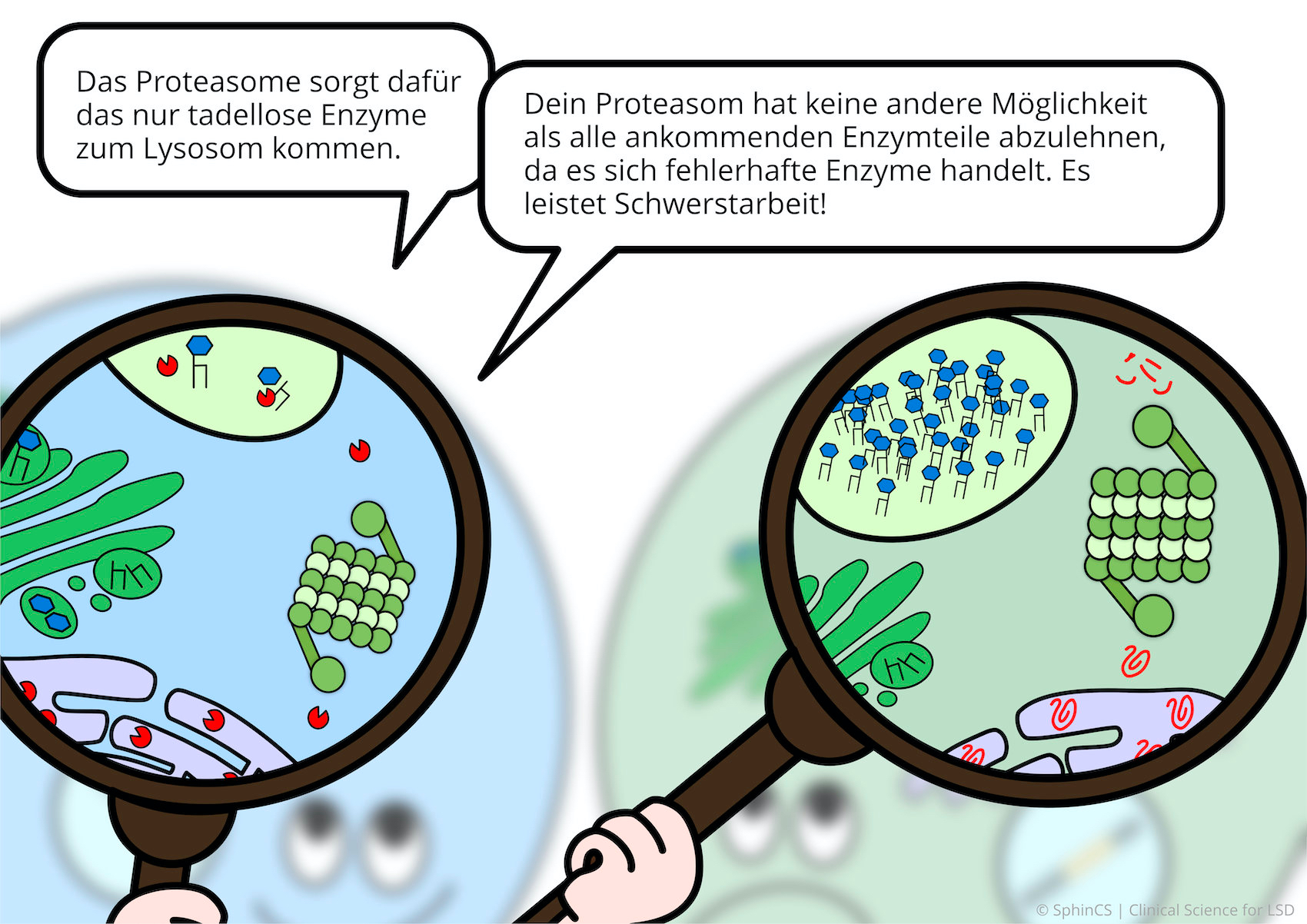
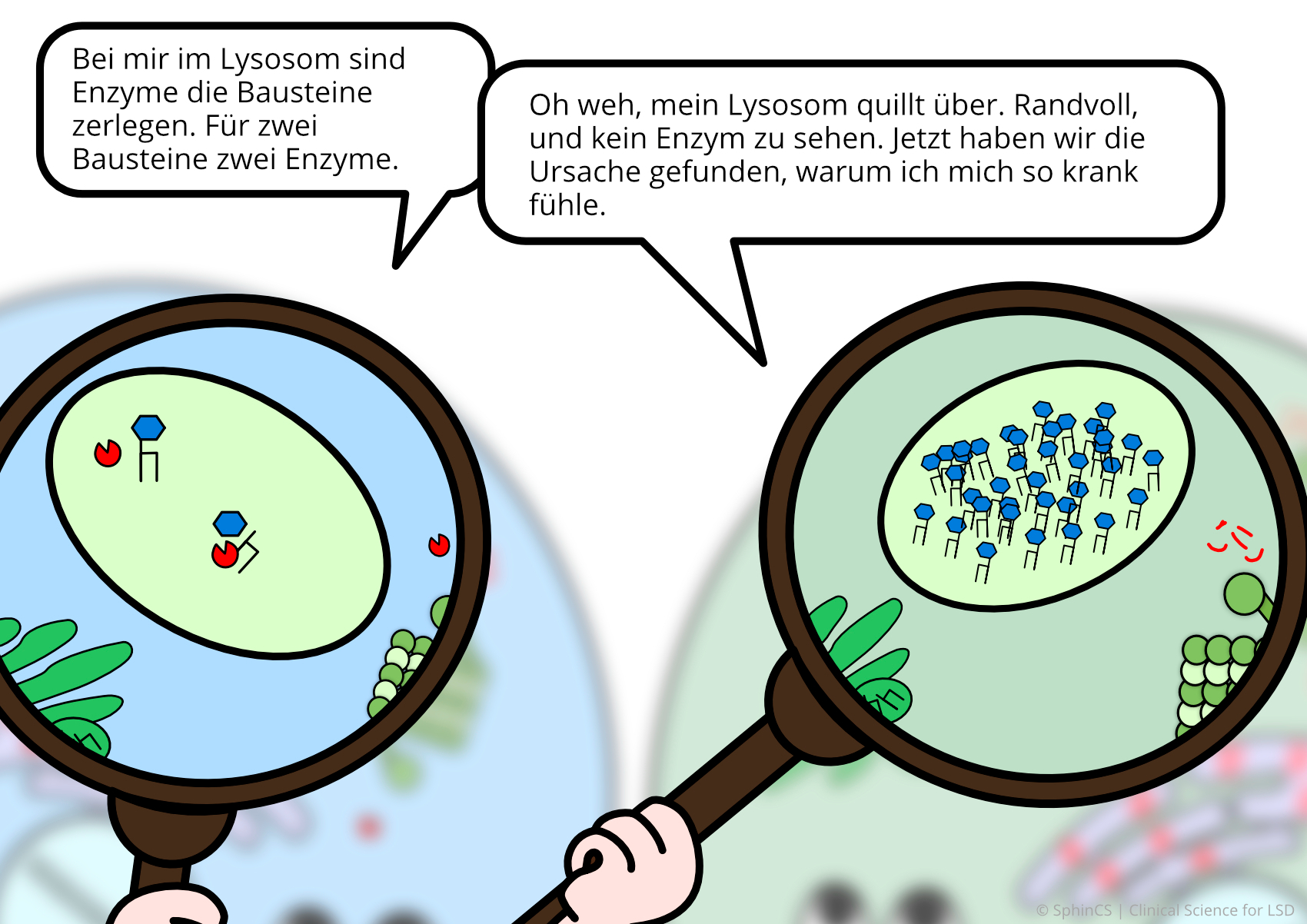



Keine Alternsbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
> 4 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
> 18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥12 and <18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
18-65 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
keine Altersbeschränkung
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
Ab 4 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
keine Alterseinschränkung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
≥18
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
16 bis 69 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
0-18 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
ab 36 Monate
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
Keine Alternsbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
> 4 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
> 18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥12 and <18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
18-65 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
keine Altersbeschränkung
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
Ab 4 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
keine Alterseinschränkung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
≥18
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
16 bis 69 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
0-18 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
ab 36 Monate
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH












