Unser Ziel ist es, die klinische Forschung bei seltenen Erkrankungen voranzubringen und für bisher unbehandelbare lysosomale Speicherkrankheiten (LSD) eine Therapie zu entwickeln. Gleichzeitig setzen wir uns für die gesundheitlichen Belange von betroffenen Patienten ein.
Wir tun dies, indem wir der wissenschaftlichen und medizinischen Gemeinschaft Einblicke in LSDs bieten und innovative Therapien mitentwickeln. Dabei sind unsere Patienten wertvolle Akteure und Partner. Wir arbeiten mit einem persönlichen Ansatz und in einer vertrauensfördernden Atmosphäre.

 Unser Lehrer, unser Mentor, unser Doktorvater: Michael Beck, den wir heute noch mit großer Freude unseren „Chef“ nannten, ist gegangen. Er hinterlässt eine große Lücke.
Unser Lehrer, unser Mentor, unser Doktorvater: Michael Beck, den wir heute noch mit großer Freude unseren „Chef“ nannten, ist gegangen. Er hinterlässt eine große Lücke.
Michael Beck arbeitete bis zu seiner Pensionierung in der Universitätsmedizin Mainz, wo er als Gründer und Leiter der Villa Metabolica weltweites Renommee für die Universitätsmedizin und die Stadt Mainz erworben hatte. In den vergangenen Jahren wirkte er am Aufbau unseres Forschungsinstituts SphinCS mit.
Von Beginn seiner beruflichen Laufbahn an widmete er sich der Betreuung schwerst kranker Kinder. Mit großer Menschlichkeit und mit viel Herzblut begleitete er betroffene Familien auf ihrem schweren Weg, gab Ihnen Hoffnung, Zuversicht und setzte sich energisch dafür ein, mögliche Behandlungsstudien unseren Patienten hier anzubieten. Zudem engagierte er sich mit großer Begeisterung in der Wissenschaft, Forschung und Lehre. Er teilte mit uns sein Wissen, begleitete uns auf unserem beruflichen und privaten Weg, förderte und unterstützte uns als Doktorvater, Chef und Freund.
Lieber Michael, wir danken Dir für Deinen unermüdlichen Einsatz und Dein Vertrauen. Wir werden Dich sehr vermissen.

Klinische Forschung für lysosomale Erkrankungen erhält glücklicherweise mehr und mehr Aufmerksamkeit. Wir möchten Ihnen helfen auf dem Laufenden zu bleiben und informieren Sie dementsprechend auch über aktuell stattfindende Studien.
Eine Übersicht über die aktuellen Therapiekonzepte mit Gen-Therapie, hämatopoetischer Stammzell-Transplantation (Knochenmarktransplantation), Enzymersatztherapie, Substrat-Reduktionstherapie und Chaperontherapie wird im Artikel "Precision Medicine for Lysosomal Disorders“ (Jul 26 2020) wissenschaftlich dargestellt. Weiterhin haben wir versucht, das Thema Therapiekonzepte bei lysosomalen Krankheiten, in einem Comic zu veranschaulichen.

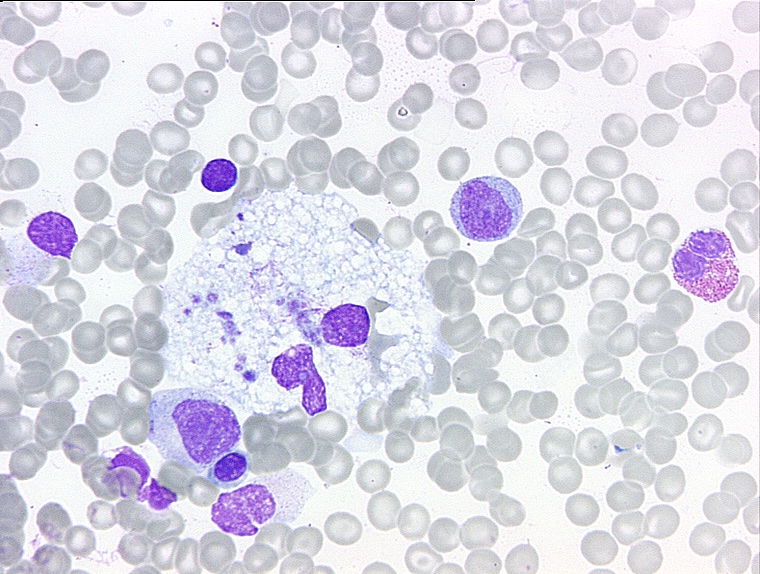
M. Gaucher ist eine autosomal-rezessiv vererbte lysosomale Speichererkrankung und wird durch die Defizienz des lysosomalen Enzyms beta-Glukozerebrosidase verursacht. Diese führt zu einer intrazellulären Speicherung von Glycosylceramid in den Zellen verschiedener Organe, vor allem in der Leber, Milz und im Knochenmark. Man unterscheidet drei Phänotypen: den M. Gaucher Typ I, Typ II und Typ III. Der M. Gaucher Typ I, der häufigste Phänotyp, bezeichnet die chronisch nicht neurologische Verlaufsform und ist charakterisiert durch eine Hepatosplenomegalie, Knochensymptome (Schmerzen, Knocheninfarkte, Osteonekrosen), Thrombozytopenie und Anämie. Der M. Gaucher Typ II ist die seltenste Verlaufsform und ist gekennzeichnet durch eine Manifestation im Säuglingsalter mit schweren rasch progressiven neurologischen Symptomen (bulbäre und pyramidale Zeichen, Myoklonische Epilepsie). Der M. Gaucher Typ III vereint neben den klinischen Zeichen des M. Gaucher Typ I progrediente neurologische Symptome (okulomotorische Apraxie, Ataxie, Epilepsie). Therapeutisch stehen 4 zugelassenen Medikamente zur Verfügung (Enzymersatztherapien mit Imiglucerase oder Velaglucerase alfa für M. Gaucher Typ I und Typ III, Substratreduktionstherapien mit Miglustat oder Eliglustat für M. Gaucher Typ I).

Lysosomale Speicherkrankheiten (Lysosomal Storage Disorders, LSD) sind eine Gruppe von mehr als 50 seltenen erblichen Stoffwechselkrankheiten. Die Krankheiten sind gekennzeichnet durch eine abnorme Anhäufung verschiedener toxischer Stoffe in den Körperzellen als Folge von Enzymmängeln.
Lysosomale Speicherkrankheiten betreffen das Lysosom, eine Struktur in den Zellen, die Stoffe wie Proteine, Kohlenhydrate und alte Zellteile abbaut, damit der Körper sie wiederverwerten kann. Infolgedessen können verschiedene Teile des Körpers betroffen sein, darunter das Skelett, das Gehirn, die Haut, das Herz und das zentrale Nervensystem. Neue lysosomale Speicherkrankheiten werden weiterhin identifiziert.