LSD are genetic, hereditary diseases that lead to the lysosomal digestion of the cell no longer functioning. The effects on humans are complex and complicated, sometimes we do not understand the attributions sufficiently.
What we do understand, however, is that unmetabolized substances accumulate in the cell and damage the cell. The cells - and consequently the organs belonging to these cells - that are damaged are very dependent on the substances that are not broken down.
In M. Pompe, for example, glycogen cannot be metabolized lysosomally in the muscle. Consequently, M. Pompe is a muscle disease.
Glycolipid - a glucosylceramide - is produced during the digestion and renewal of blood cells, especially in the spleen and bone marrow. In M. Gaucher, glucosylceramide is not metabolized. It accumulates in the macrophages of the spleen and bone marrow. Spleen enlargement and infiltration of the bone marrow are the most important findings in Gaucher's disease.
Further information on diagnostics and our diagnostic service.
When diagnosing storage disease, four questions must be addressed:

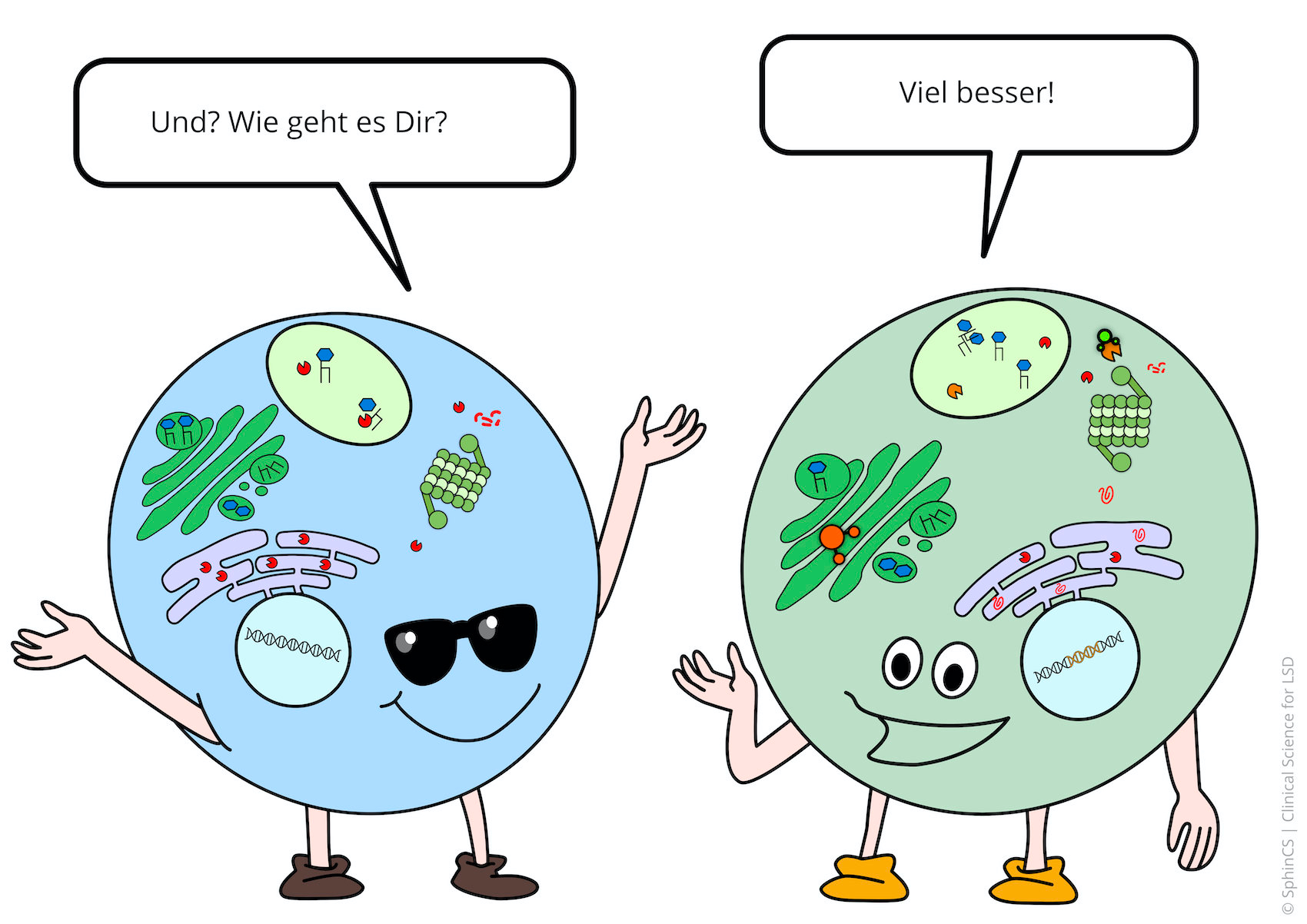
[Test is machine translated from German language] Treatment not possible, rare, complex, difficult to understand, poor prognosis, ... these are the keywords that have stuck from medical school. But if we summarize current 2020 research, there are already effective and approved therapies and further therapeutic approaches give hope.
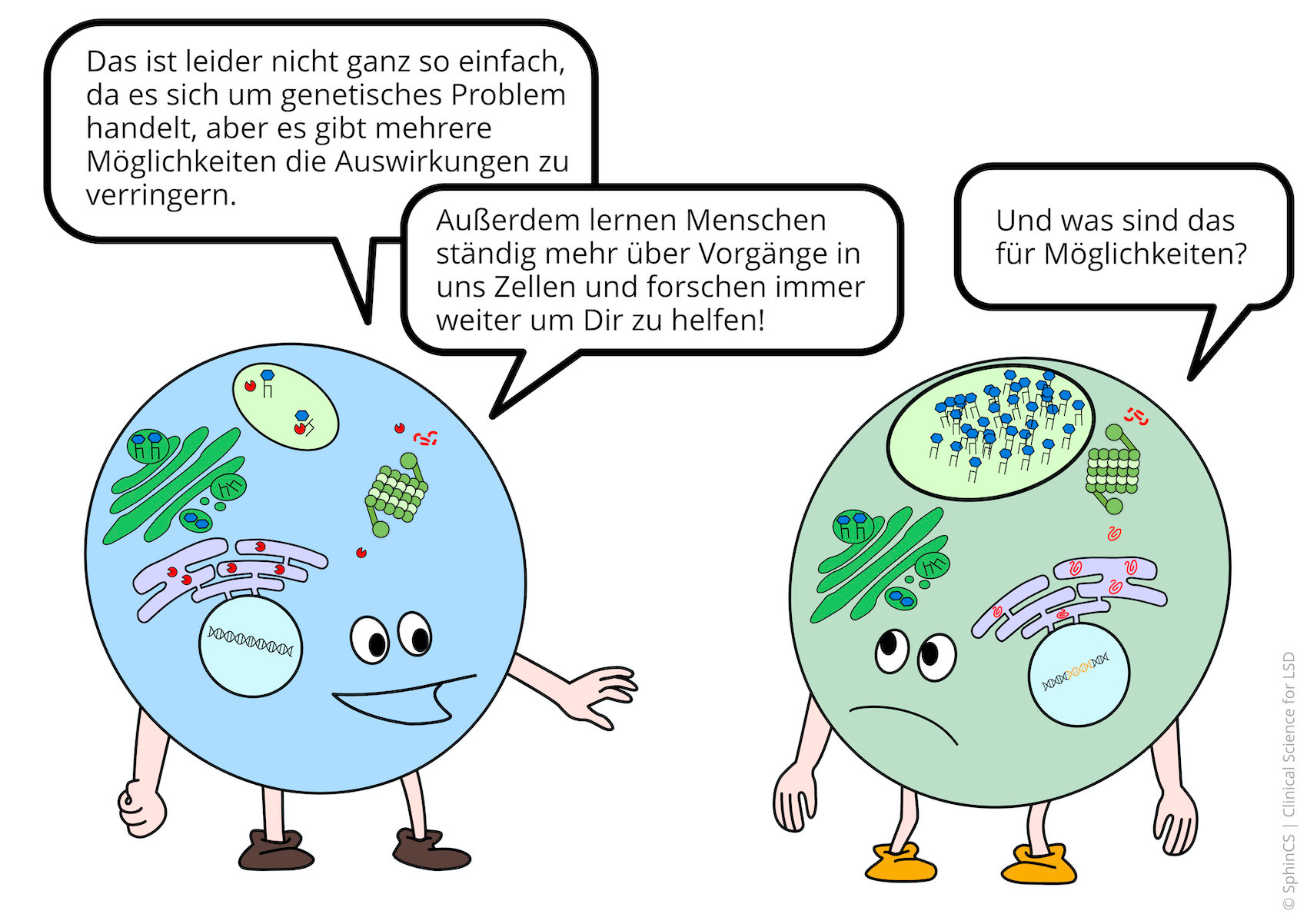
Treatment of the central nervous system is a particular challenge that continues to evolve through substrate inhibition and gene therapy
As understanding of what happens in cells and organs in lysosomal diseases improves, basic research continues to develop new targets for therapies
In 1983, the first bone marrow transplantation was performed for a lysosomal disease. A boy with MPS 1 was transplanted in London by Dr. Hobbs. It has not been proven that all lysosomal diseases can be treated in this way. It is the severe courses of a few diseases, such as MPS 1 and Farber's disease, for which bone marrow transplantation remains the treatment of choice in 2020.
In 1991, a murmur went through the medical press. Roscoe Brady and his team at the NIH had treated 12 Gaucher patients in a study with the very enzyme ( glucocerebrosidase ) that is missing in this disease. The enzyme was given via infusions 14 days a week. The disease symptoms spleen enlargement deficiency of platelets and bleeding tendency responded excellently to the therapy. At that time, the enzyme was obtained from placenta, but nowadays it is produced biotechnologically in larger quantities and more safely.
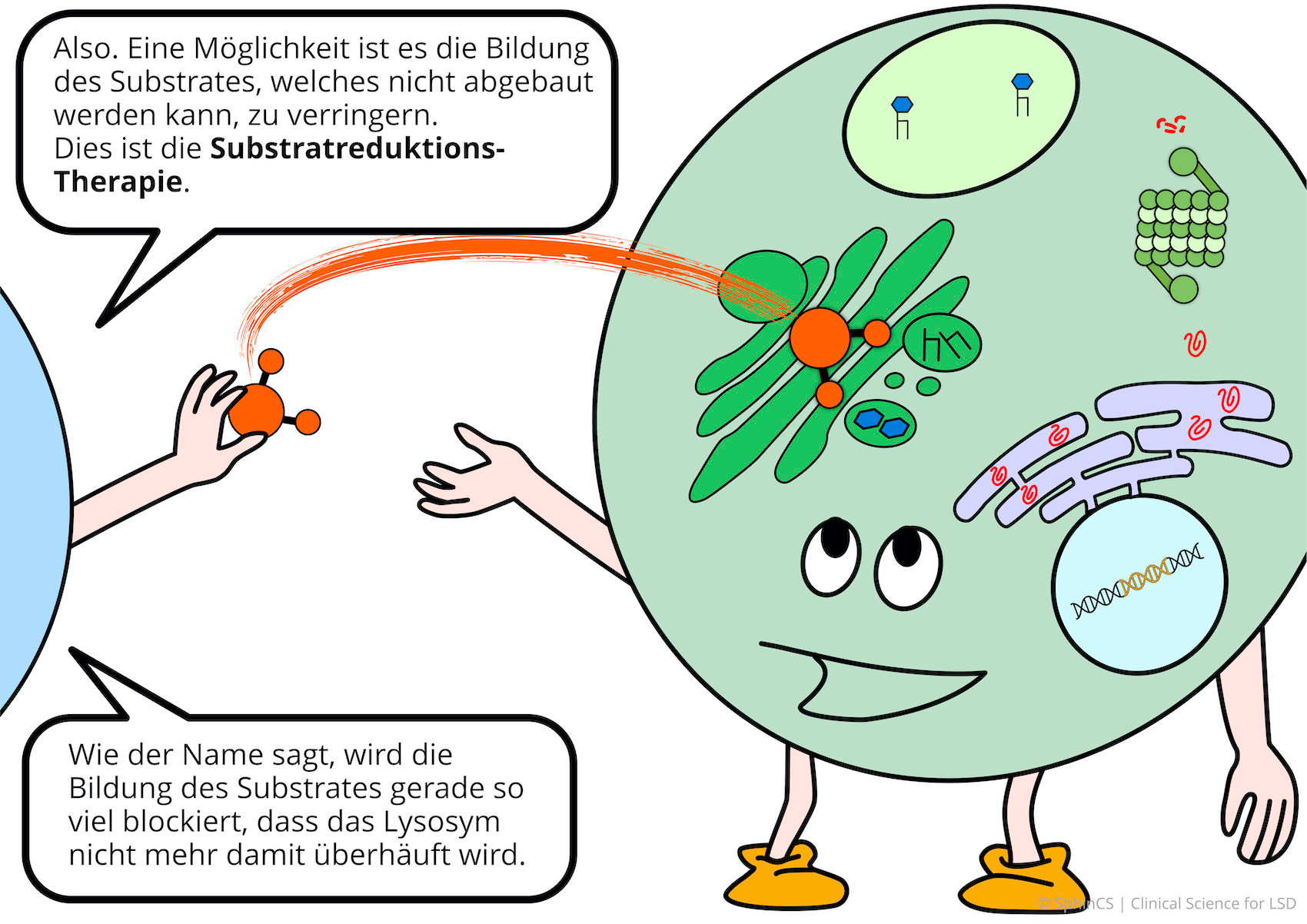
The principle of therapy is based on the fact that the substance that cannot be degraded (=substrate) is only produced to a very limited extent. The substrate-building enzyme is inhibited, then the lack of the enzyme that breaks down the substrate is hardly noticeable.
Chaperones bind specifically to misfolded enzymes and stabilize the structure of the enzyme. The type of mutation can be used to determine whether a mutation causes misfolding. Patients with such mutations are eligible for chaperone therapy.
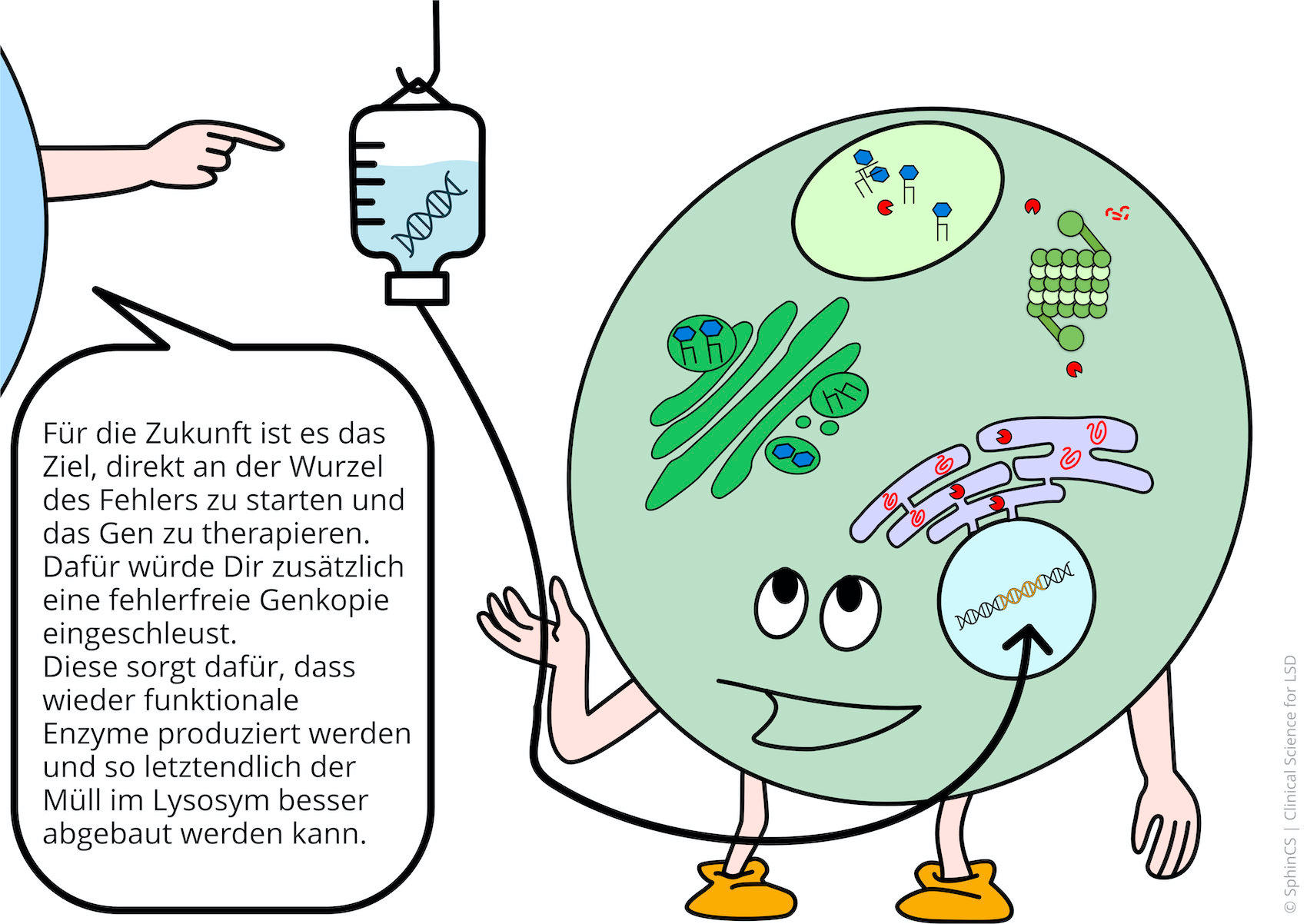
Gene therapies cure inherited diseases by introducing a defect-free gene copy. Sounds simple, but it is highly complicated. The introduction is achieved by viral vectors. In particular, successful basic research with the AAV9 vector, which introduces the healthy gene into nerve cells, has led to the development of numerous therapy studies for lysosomal diseases in the last two years.
For a different kind of overview, we invite you to look at the following comic. Just click or tap on the first image or download the comic as a PDF.

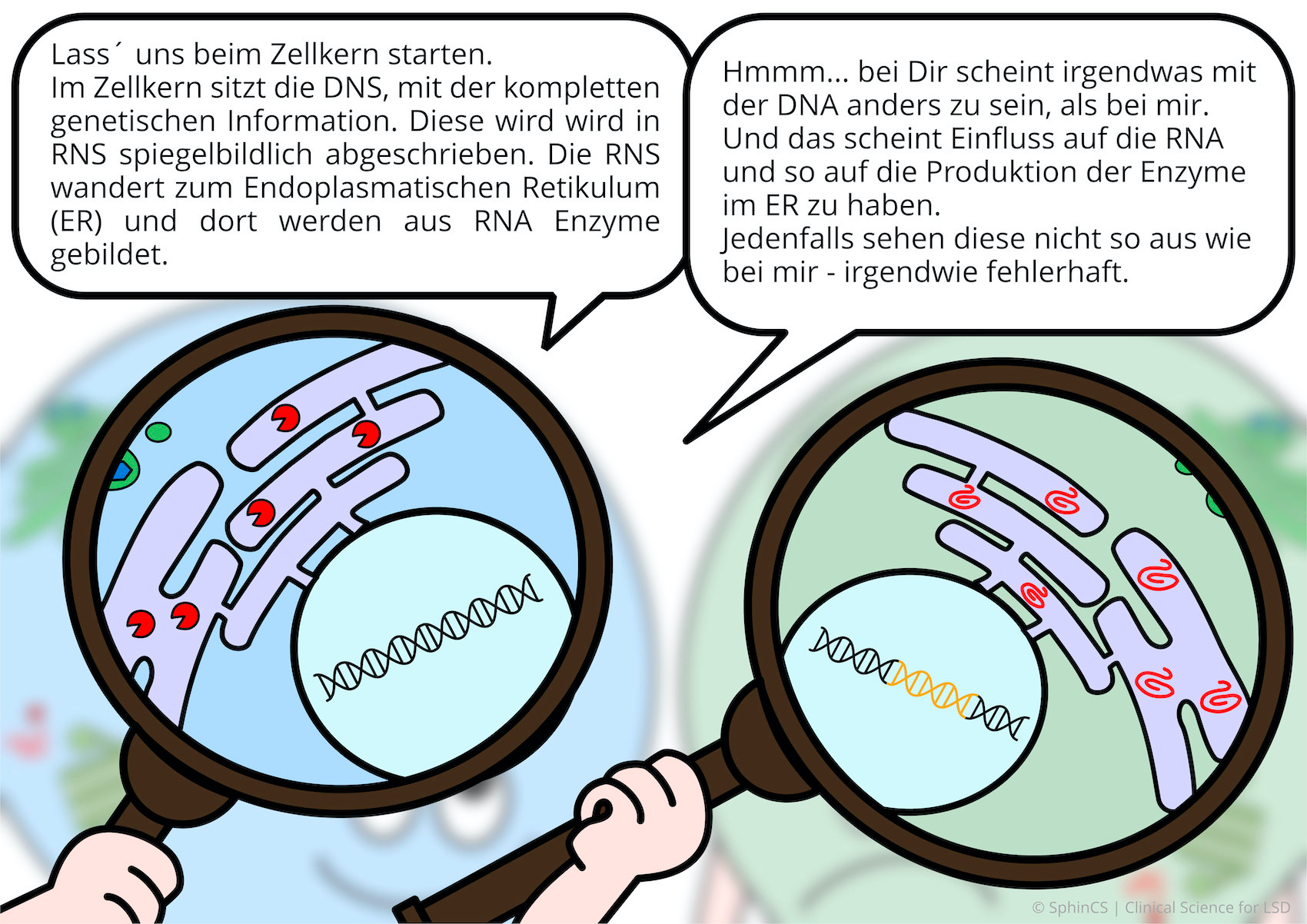
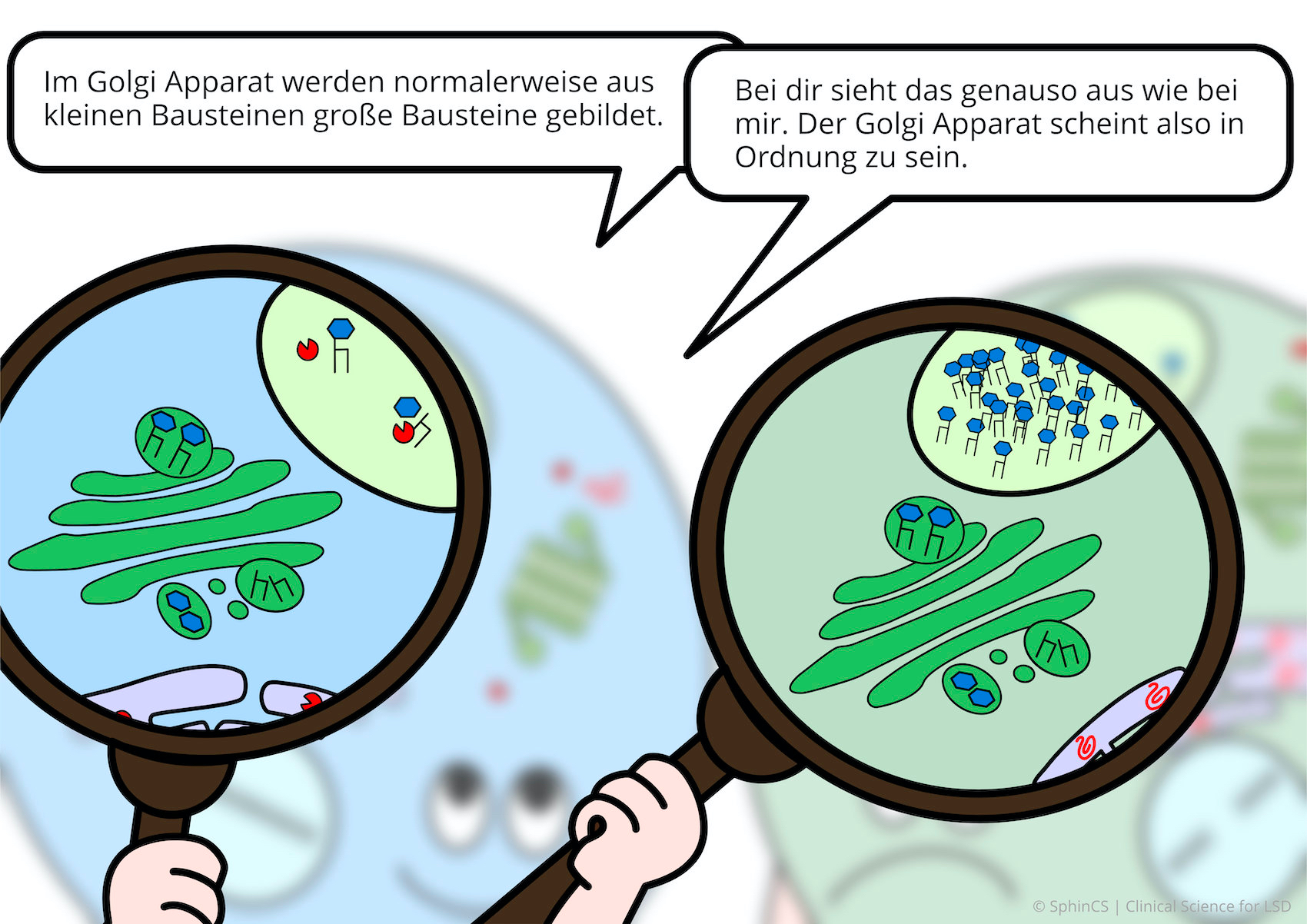
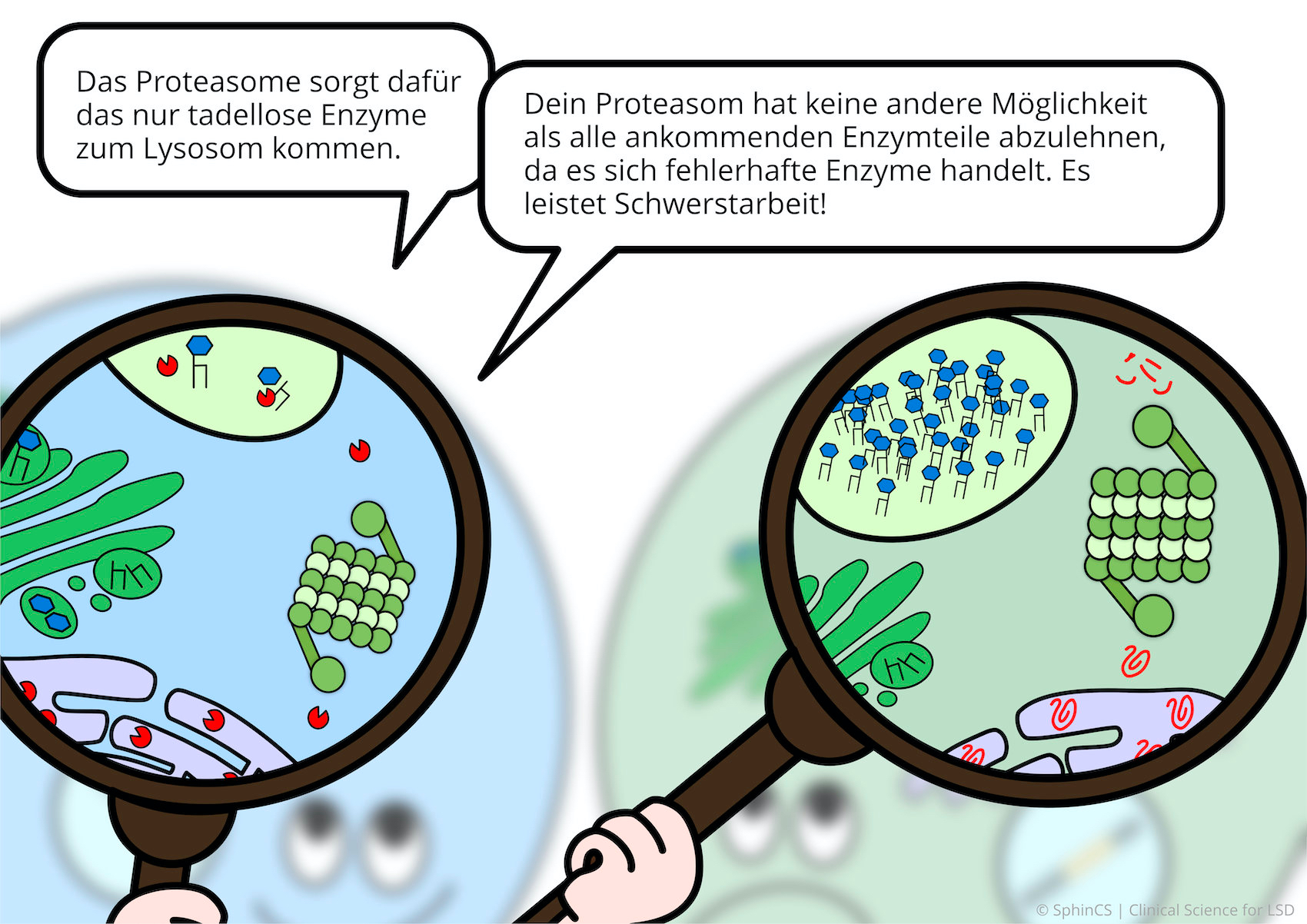
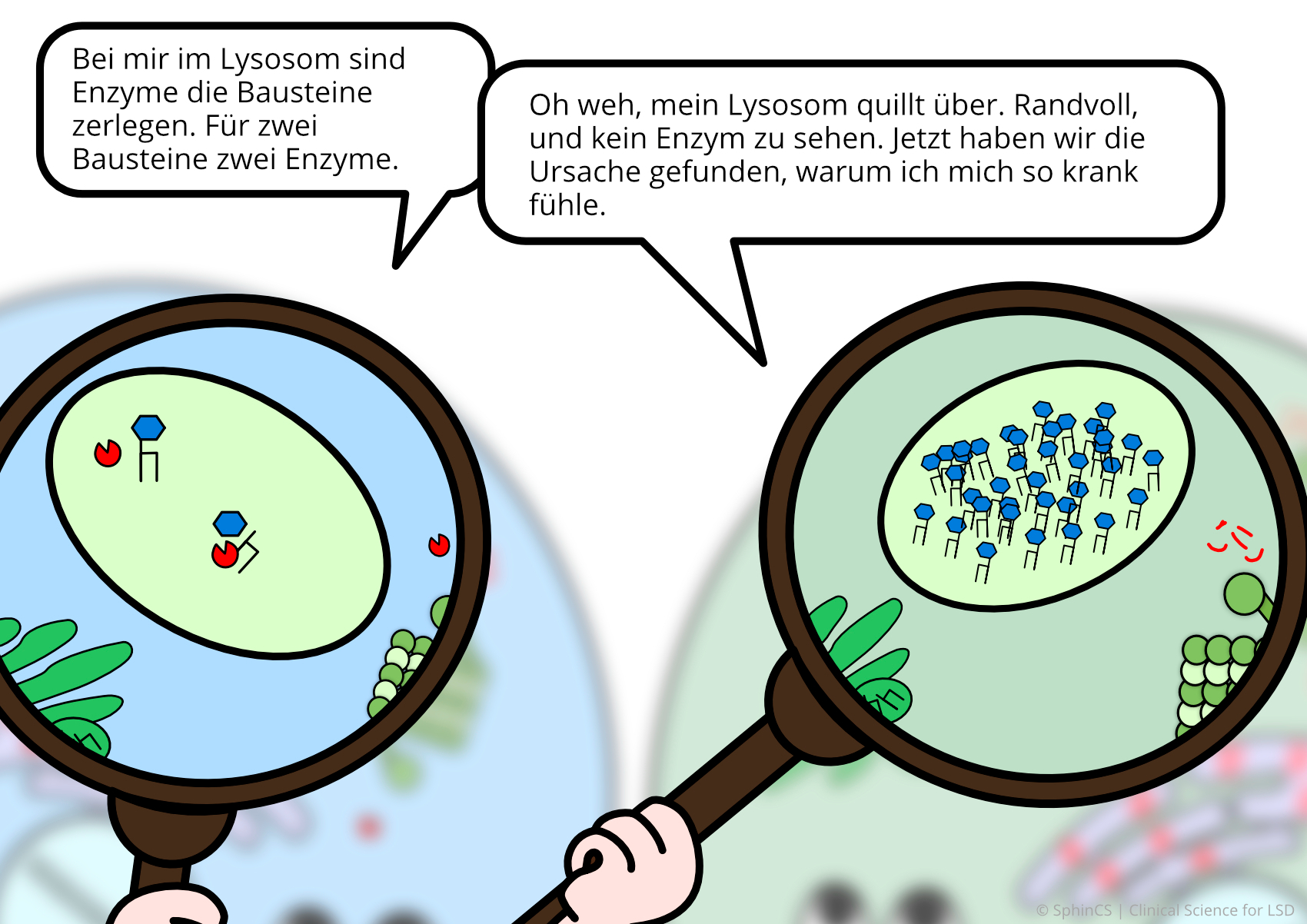



No age limit
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
> 4 Years
Status active
Patient admission recruiting
Institution SphinCS GmbH
No age limit
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
No age limit
Status active
Patient admission recruiting
Institution SphinCS GmbH
> 18 Years
Status active
Patient admission closed
Institution SphinCS GmbH
≥12 and <18 years
Status active
Patient admission closed
Institution SphinCS GmbH
18-65 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
without age restriction
Status active
Patient admission closed
Institution SphinCS GmbH
From 4 years ago
Status active
Patient admission closed
Institution SphinCS GmbH
no age restriction
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
≥18
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
16 to 69 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
0-18 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
≥ 2 to <26 years
Status active
Patient admission closed
Institution SphinCS GmbH
≥ 2 to <26 years
Status active
Patient admission closed
Institution SphinCS GmbH
from 36 months
Status active
Patient admission recruiting
Institution SphinCS GmbH
Keine Alternsbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
> 4 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
Keine Altersbegrenzung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
> 18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥12 and <18 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
18-65 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
keine Altersbeschränkung
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
Ab 4 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
keine Alterseinschränkung
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
≥18
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
16 bis 69 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
0-18 Jahre
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
≥ 2 bis <26 Jahre
Status Aktiv
Patientenaufnahme abgeschlossen
Institution SphinCS GmbH
ab 36 Monate
Status Aktiv
Patientenaufnahme rekrutierend
Institution SphinCS GmbH











