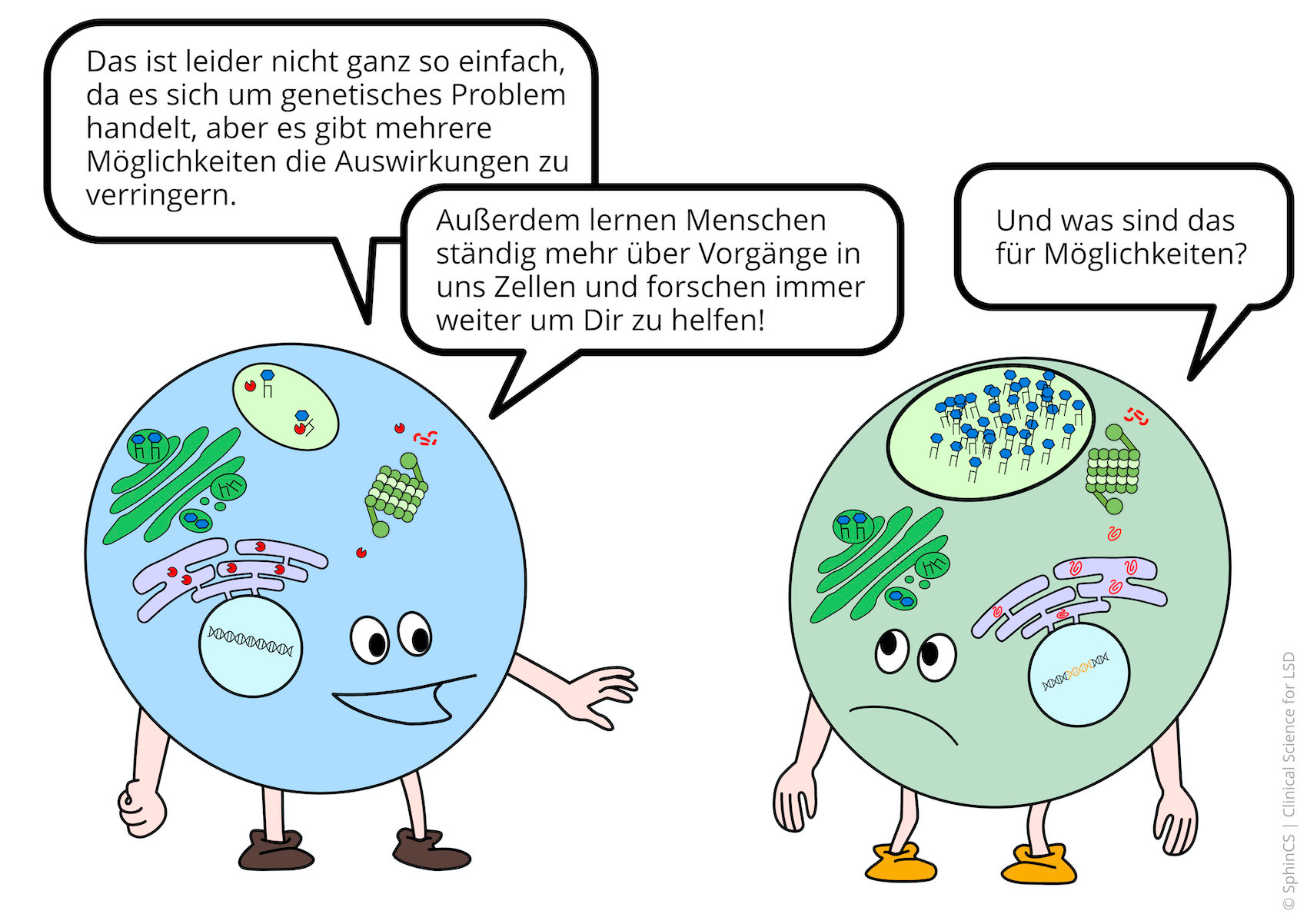
LSD are genetic, hereditary diseases that lead to the lysosomal digestion of the cell no longer functioning. The effects on humans are complex and complicated, sometimes we do not understand the attributions sufficiently.
What we do understand, however, is that unmetabolized substances accumulate in the cell and damage the cell. The cells - and consequently the organs belonging to these cells - that are damaged are very dependent on the substances that are not broken down.
In M. Pompe, for example, glycogen cannot be metabolized lysosomally in the muscle. Consequently, M. Pompe is a muscle disease.
Glycolipid - a glucosylceramide - is produced during the digestion and renewal of blood cells, especially in the spleen and bone marrow. In M. Gaucher, glucosylceramide is not metabolized. It accumulates in the macrophages of the spleen and bone marrow. Spleen enlargement and infiltration of the bone marrow are the most important findings in Gaucher's disease.
Further information on diagnostics and our diagnostic service.
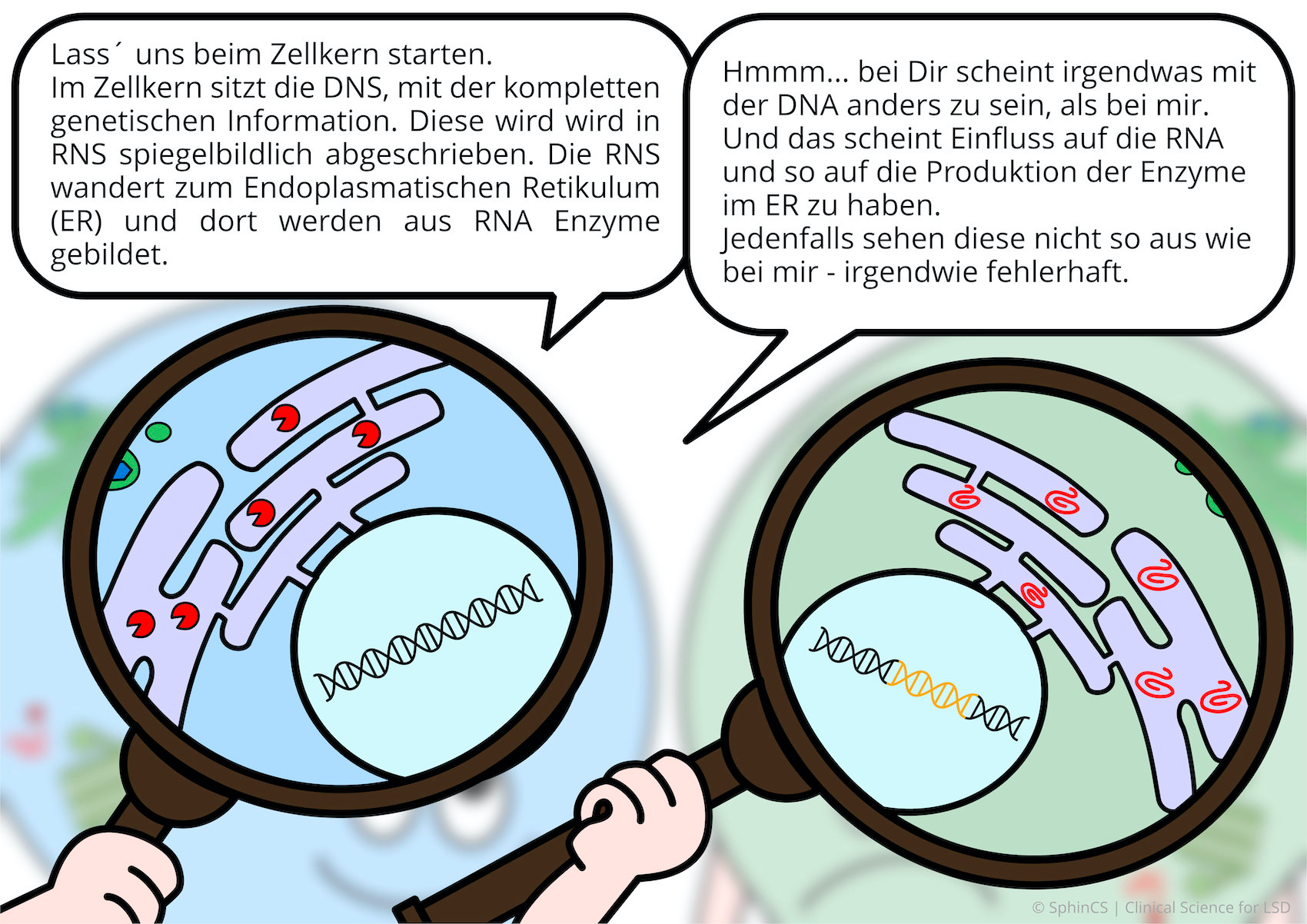
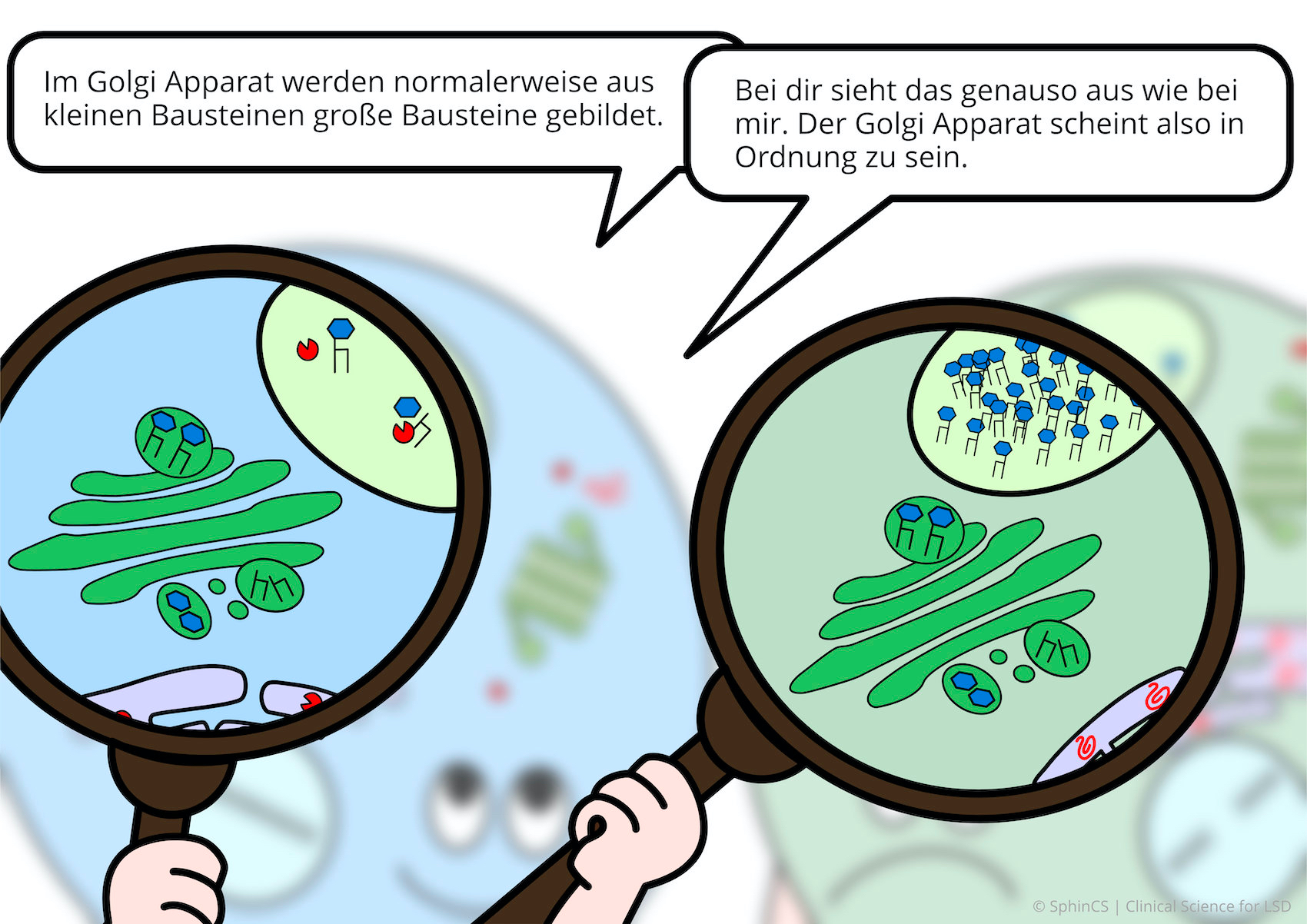
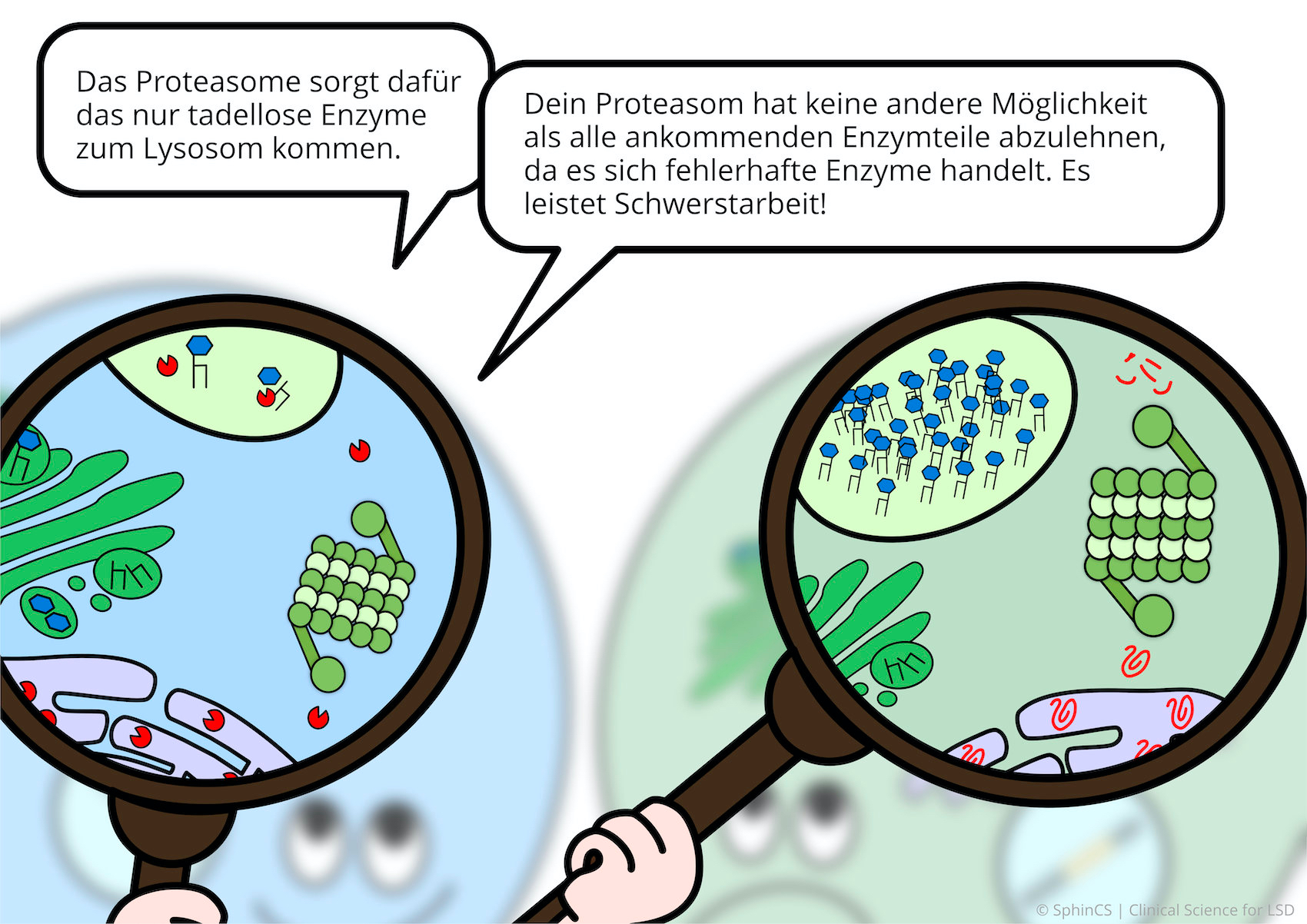
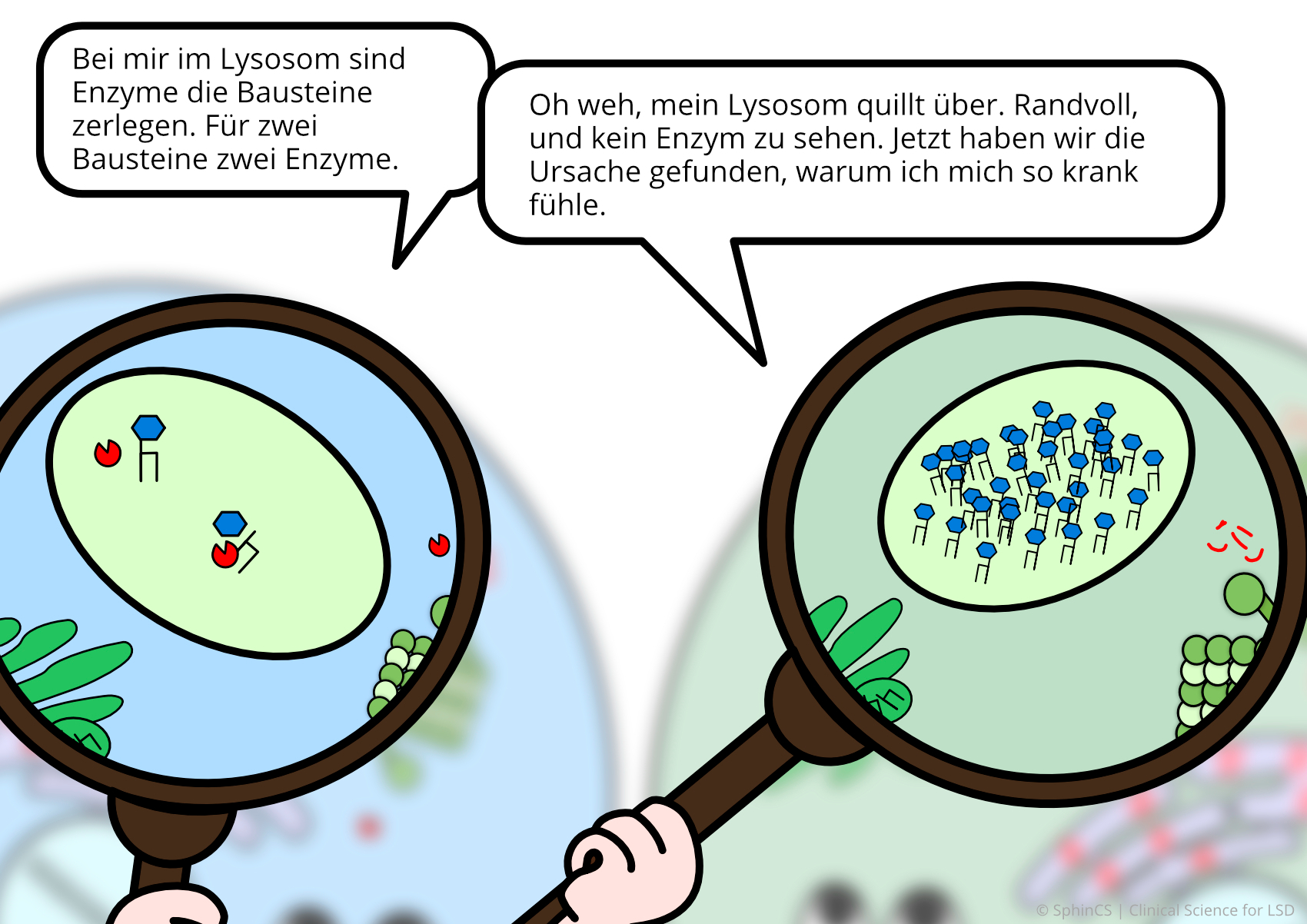
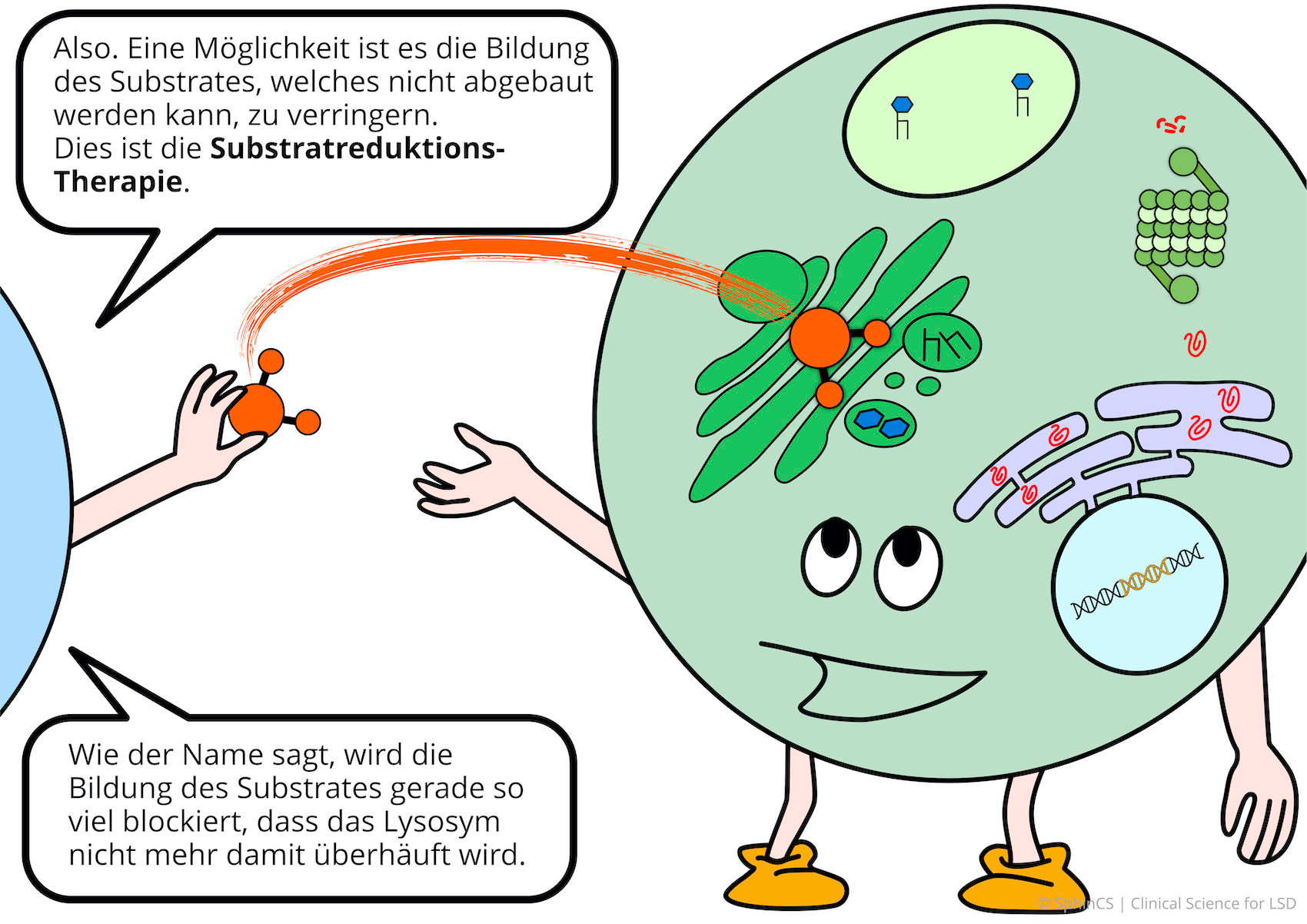
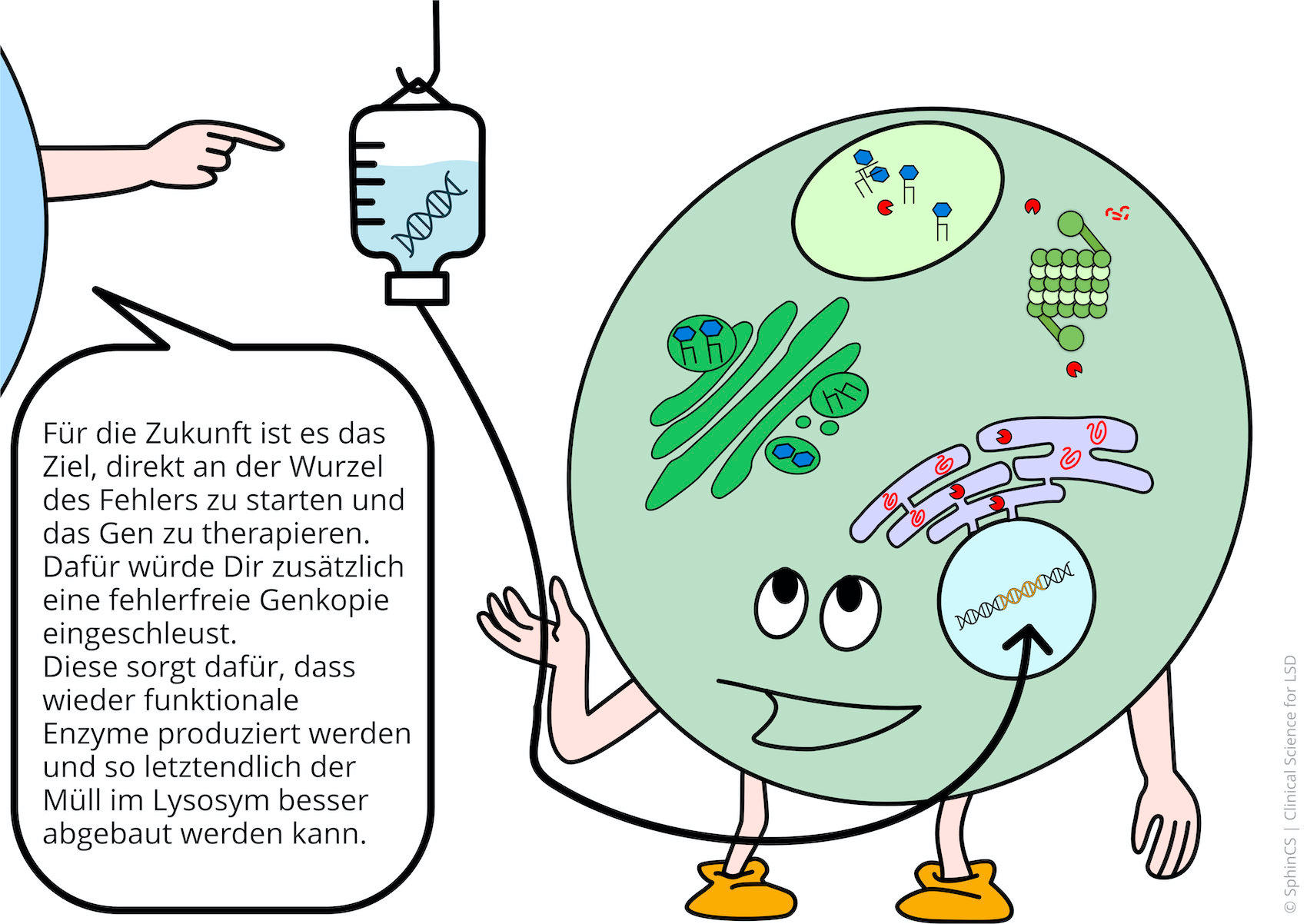
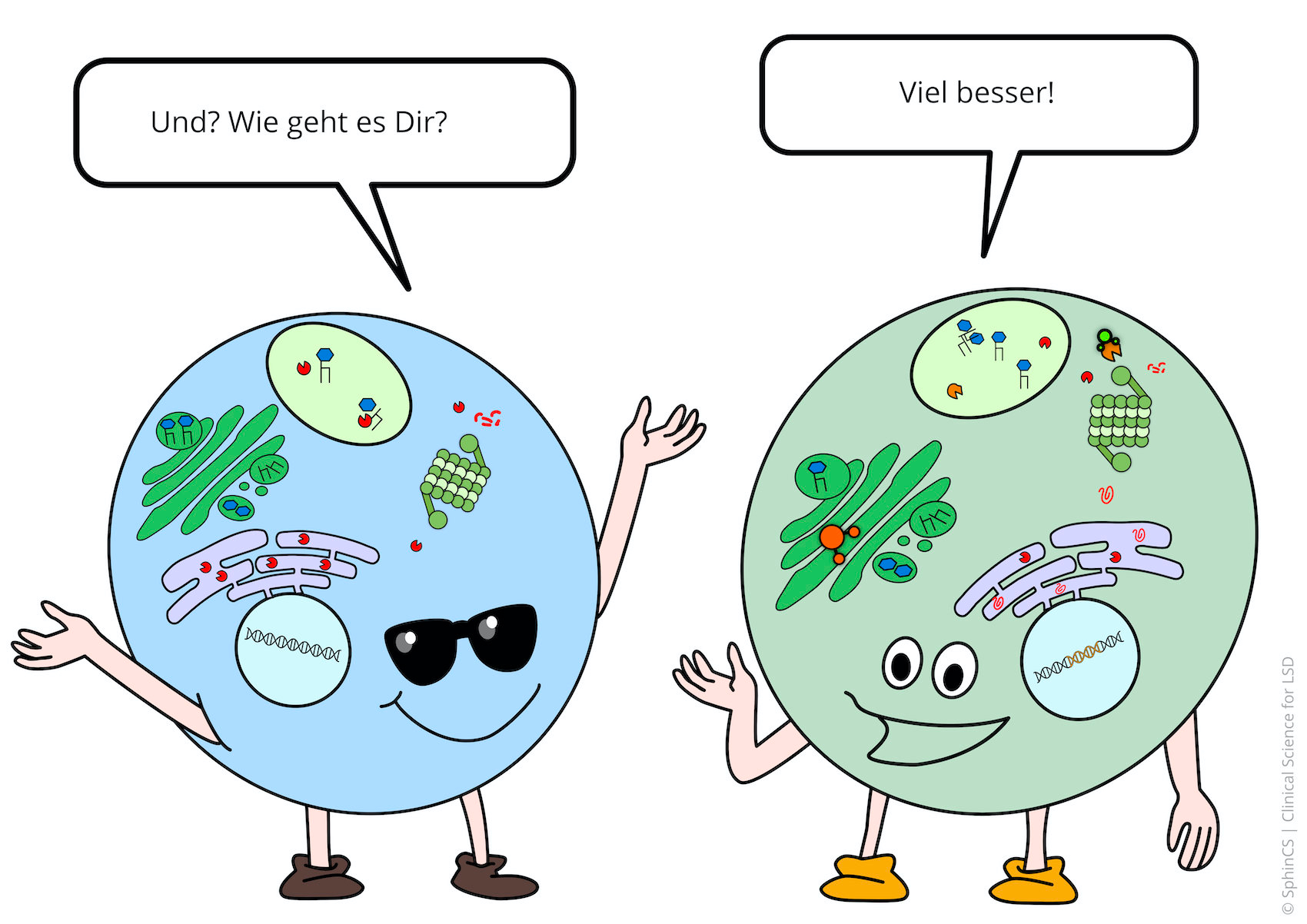
Nowadays there are several possibilities of therapy available for the different LSD. For a first overview, we invite you to have a look at the following comic. Just click or tap on the first picture or download the comic as a PDF.




No age limit
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
> 4 Years
Status active
Patient admission recruiting
Institution SphinCS GmbH
No age limit
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
No age limit
Status active
Patient admission recruiting
Institution SphinCS GmbH
> 18 Years
Status active
Patient admission closed
Institution SphinCS GmbH
≥12 and <18 years
Status active
Patient admission closed
Institution SphinCS GmbH
18-65 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
without age restriction
Status active
Patient admission closed
Institution SphinCS GmbH
From 4 years ago
Status active
Patient admission closed
Institution SphinCS GmbH
no age restriction
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
≥18
Status active
Patient admission recruiting
Institution SphinCS Lyso gemeinnützige UG (haftungsbeschränkt)
16 to 69 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
0-18 years
Status active
Patient admission recruiting
Institution SphinCS GmbH
≥ 2 to <26 years
Status active
Patient admission closed
Institution SphinCS GmbH
≥ 2 to <26 years
Status active
Patient admission closed
Institution SphinCS GmbH
from 36 months
Status active
Patient admission recruiting
Institution SphinCS GmbH











